Langer–Giedion sindromu (LGS) - Çox nadir bir avtosomal dominant irsi xəstəlik. Xromosom 8-dəki kiçik bir materialın silinməsi ilə yaranır. Bu sindrom 1960-cı illərdə bu vəziyyət üzərində əsas tədqiqatları aparan iki həkimin adı ilə adlandırılıb. Diaqnoz adətən doğuşda və ya erkən uşaqlıq dövründə qoyulur.
Əlamətlər və simptomlar
Bu xəstəliklə əlaqəli olan xüsusiyyətlərə yüngül və ya orta dərəcəli öyrənmə çətinlikləri, qısa boy, unikal üz xüsusiyyətləri, kiçik baş və sümük anormalıqları, o cümlədən sümük səthlərindən çıxan sümük böyümələri daxildir.
Kraniyofasial
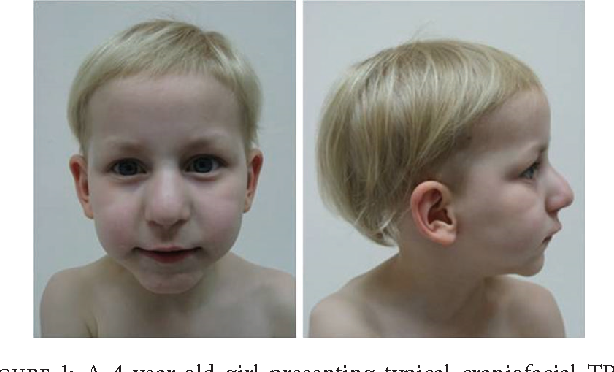
Langer–Giedion sindromu olan şəxslər tipik kraniyofasial anomaliyalar göstərə bilərlər. Bunlar arasında uzun və seçilən filtrum, incə üst dodaq, geniş aralıqlı gözlər, şişkin burun ucu, geniş burun körpüsü, geniş burun dəlikləri, mikrognatya, retrognatya, dərin yerləşmiş gözlər və iri qulaqlar vardır. Baş, adətən, eyni yaşda və cinsdə olan sağlam fərdlərə nisbətən çox kiçikdir. Diş anomaliyaları, məsələn, supernumerar mərkəzi incisivlər və bəzi dişlərin olmaması meydana çıxa bilər.
Muskuloskeletal
Langer–Giedion sindromu olan bir şəxsin sağ ayağında tipik xüsusiyyətlər Langer–Giedion sindromu, əllərin falanjalarının konus şəklində epifizlərini və qısa barmaqları və barmaq uclarını yaradır. Beşinci barmaqlar bəzən əyilir. Əllər və ayaqlar xaricində də sümük anomaliyaları meydana çıxa bilər. Bunlar arasında qanad şəkilli skapula, nazik qabırğalar və skolioz yer alır. Bundan əlavə, Langer–Giedion sindromu olan fərdlər, tədricən bud sümüklərinin başında degenerasiya kimi "Legg–Calvé–Perthes" xəstəliyində müşahidə olunan problemləri inkişaf etdirə bilərlər.
Xüsusiyyətlər
Langer–Giedion sindromu ilkin olaraq oynaq hipermobilikliyinə səbəb olur. Bu, uşaqlıq dövrü ilə orta uşaq yaşları arasında osteokondromalar inkişaf etməyə başladıqda, həyatın irəliləyən dövrlərində oynaq sərtliyinə çevrilir, bu da hərəkət qabiliyyətini azaldır. Bud displaziyası mövcud ola bilər və adətən gənc yaşda inkişaf edir, amma körpəlik və ya uşaqlıqda da baş verə bilər. Ektodermal displaziya, Langer–Giedion sindromunun əsas xüsusiyyətlərindən biridir.
Langer–Giedion sindromu olan şəxslərin əksəriyyətində saç azlığı müşahidə edilir. Bu xüsusiyyət kişilərdə daha çox nəzərə çarpır, çünki onlar tez-tez "pubertadan" sonra "alopecia" yaşayırlar. Buna baxmayaraq, qaşlar bəzən qeyri-adi dərəcədə qalındır.
Səbəb
Sindrom xromosom 8-in uzun qolunun kiçik bir hissəsinin, bir çox genləri ehtiva edən bir hissəsinin itirilməsi nəticəsində yaranır. Bu genlərin itməsi Langer–Giedion sindromunun ümumi xüsusiyyətlərinin bəzilərinə səbəb olur. İtirilmiş hissə xromosom 8-də "8q23.2–q24.1" arasındadır. Bu region "TRPS1" və "EXT1" genlərini əhatə edir.
Diaqnoz
Diaqnoz klinik tapıntılara əsaslanır və sitogenetik testlə təsdiqlənə bilər, burada silinmə təxminən 5 Mb (milyonlarca baza cütü) təşkil edir. Hazırda, bu genetik bölgənin itirilmə dərəcəsini və silinmiş genləri müəyyən etmək üçün xəstənin periferik qanında "aCGH" (array chromosome hybridization genome) tədqiqatı aparmaq adi bir tətbiqdir.
Müalicə
Heç bir genetik sindrom müalicə oluna bilməz, amma bəzi simptomlar üçün müalicələr mövcuddur. Eksternal fiksatorlar, üz və əzələ quruluşlarının bərpası üçün istifadə edilmişdir.
İstinadlar
https://pmc.ncbi.nlm.nih.gov/articles/PMC5006835/
https://en.wikipedia.org/wiki/Langer%E2%80%93Giedion_syndrome
https://www.orpha.net/en/disease/detail/502
https://medlineplus.gov/genetics/condition/trichorhinophalangeal-syndrome-type-ii/
https://rarediseases.org/rare-diseases/trichorhinophalangeal-syndrome-type-ii/
https://nfed.org/learn/types/trichorhinophalangeal-syndrome-2/
https://pacs.de/term/langer-giedion-syndrome_2
https://www.encyclopedia.com/science/encyclopedias-almanacs-transcripts-and-maps/langer-giedion-syndrome
https://www.wikidoc.org/index.php/Langer-Giedion_syndrome
https://bugsigdb.org/Langer-Giedion_syndrome
https://karger.com/cgr/article-abstract/162/1-2/46/822013/Minimal-Critical-Region-and-Genes-for-a-Typical?redirectedFrom=PDF
https://www.reumatologiaclinica.org/en-trichorhinophalangeal-syndrome-type-1-giedion-articulo-S1699258X2200184X
Tarix : 5 fevral 2025